Drug Design with Computers
How Do We Turn a Protein into a Medicine?
Lecture 1 — From Protein to Drug Target
You already know more than you think
| You already know | We will build on this |
|---|---|
| Proteins are chains of amino acids | The sequence folds into a precise 3D shape |
| Enzymes have active sites | The shape of that site determines which molecules can bind |
| Inhibitors block enzyme activity | We can design molecules to be better inhibitors |
| Mutations change protein sequence | A single amino acid change can make a drug stop working |
One question drives following steps
How do we design a molecule to switch it off?"
- Which protein do we target?
- Where exactly on that protein does the molecule need to bind?
- What shape and chemistry does the molecule need to have?
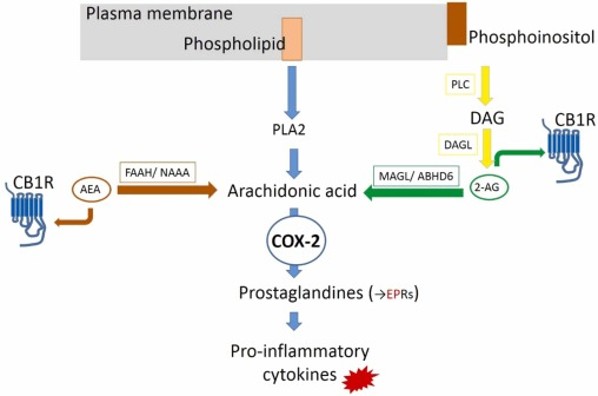
COX reaction pathway
The sequence is the blueprint
the shape is the machine
"The same protein chain always folds into the same 3D shape. That shape determines everything the protein can do — including whether a drug can bind to it."
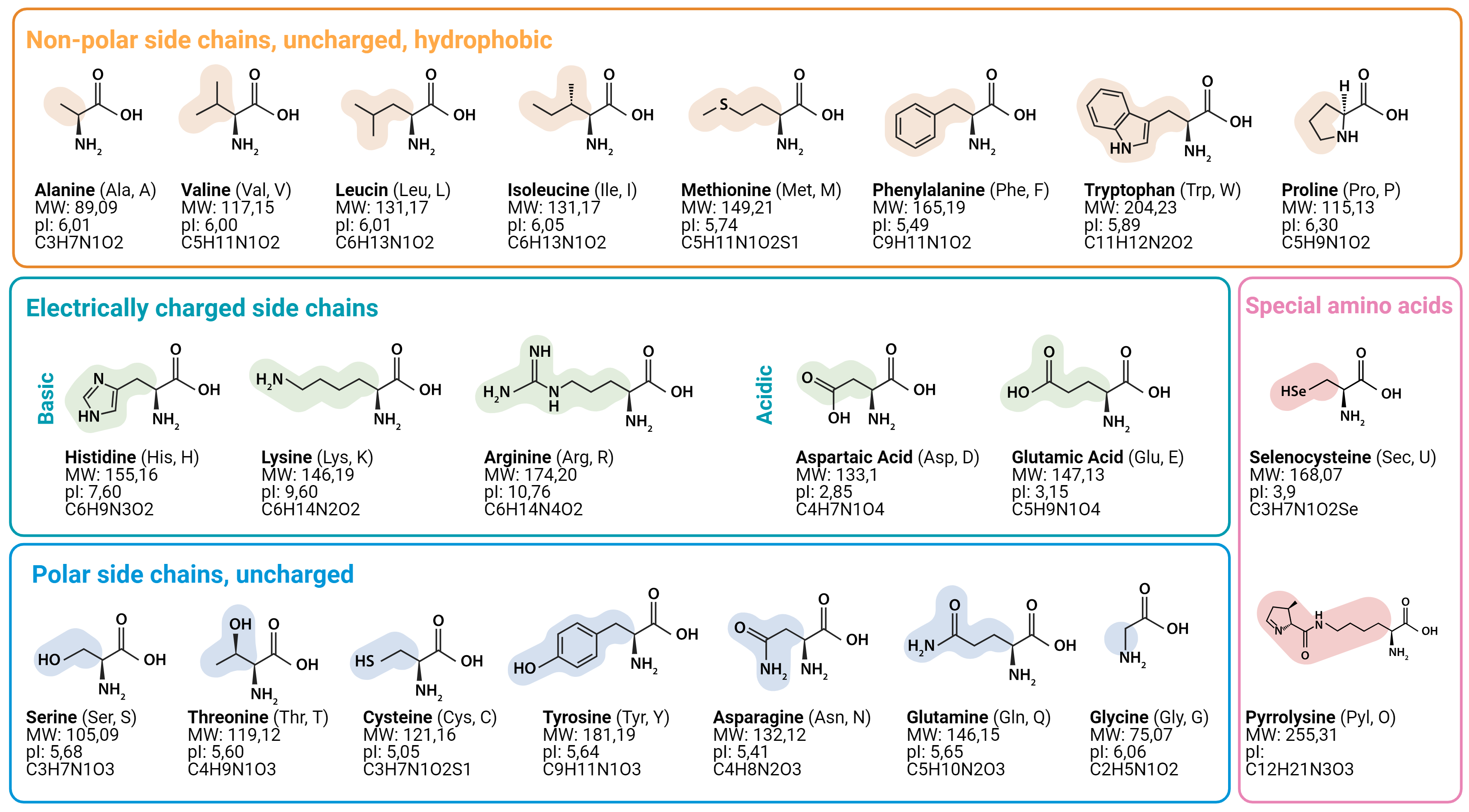
The 20 amino acids have very different personalities
| Amino acid | Property | Drug design relevance |
|---|---|---|
| Alanine (Ala, A) | Tiny, non-polar | Used as a probe in alanine scanning — its side chain does almost nothing |
| Phenylalanine (Phe, F) | Large, hydrophobic aromatic ring | Likes to sit in hydrophobic pockets — often lines binding sites |
| Arginine (Arg, R) | Positively charged | Forms strong interactions with negatively charged drug molecules |
| Serine (Ser, S) | Polar, has -OH group | Forms hydrogen bonds — important for aspirin's covalent binding to COX-2 |
Side chains are what make each amino acid different.
It's the side chains that determine how a drug binds.
Disease often begins with a protein doing its job too well
Normal State:
Stimulus → Enzyme active → Normal amount of product → Healthy outcome
Disease State:
Overactivation → Enzyme too active → Too much product → Inflammation / infection
Example 1: COX-2 & Inflammation
COX-2 converts arachidonic acid into prostaglandins (causes pain). Overproduction = chronic inflammation. This is the target of ibuprofen (Projects 3 & 4).
Example 2: β-Lactamase & Resistance
Bacteria produce an enzyme that destroys penicillin. Natural selection favours bacteria with this enzyme (Project 6).
Not every protein is a good drug target
-
1. Essential to the disease process.
If you switch it off, the disease gets better. (e.g., Blocking COX-2 relieves pain). -
2. Has a binding pocket the drug can physically fit into.
Many proteins are flat. Good targets have a deep, well-defined cavity (like the COX-2 hydrophobic channel). -
3. Different enough from healthy human proteins.
Prevents serious side effects. Bacteria have β-Lactamase; humans don't.
Drugs work by getting in the way
Competitive Inhibition
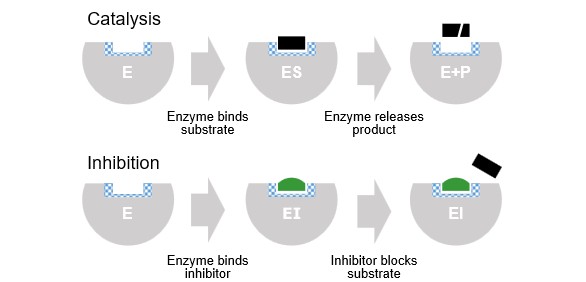
Non-competitive Inhibition
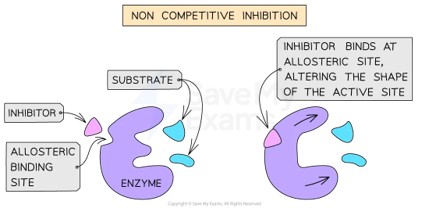
Both types work by changing the geometry. 3D shape matters — a drug that's the wrong shape simply doesn't fit!
The Protein Data Bank (PDB)
The world's protein library
A global, open-access database of experimentally determined protein structures (>220,000 structures as of 2025). Every structure contains the 3D coordinates of every atom.
- Unique 4-character ID (e.g.,
1LYZfor Lysozyme,4PH9for COX-2) - 3D coordinates (x, y, z) of every atom
- The amino acid sequence & Resolution (Å)
- Bound ligands (drugs, cofactors)
Resolution: How sharp is the picture?
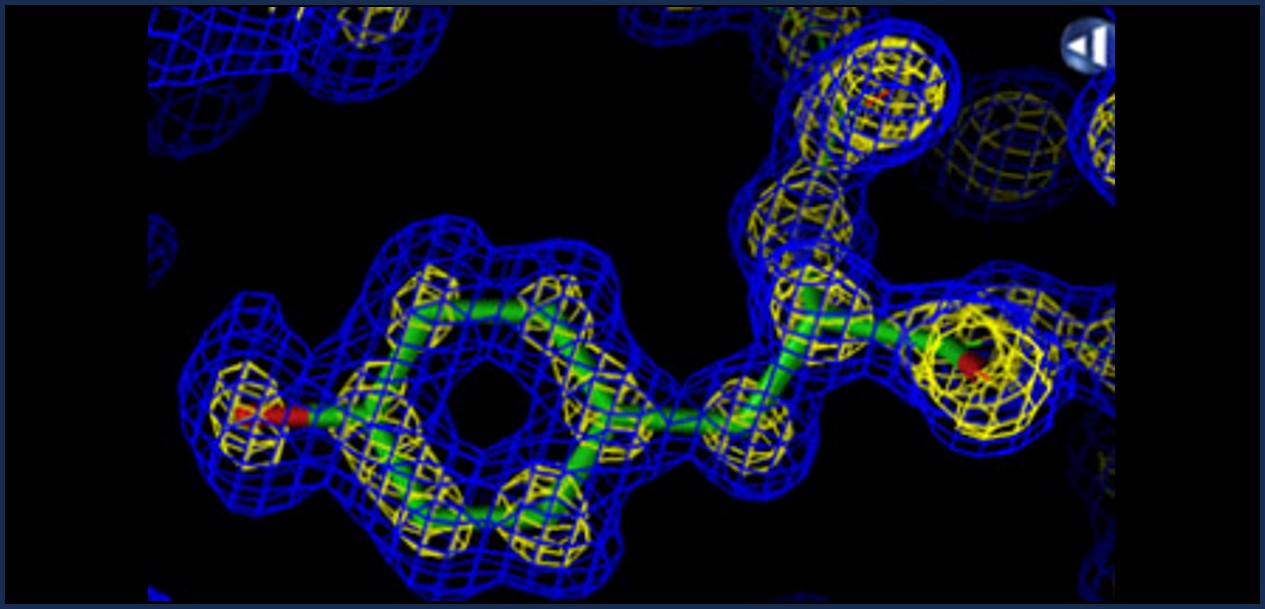
1.0 Å — Every atom precise
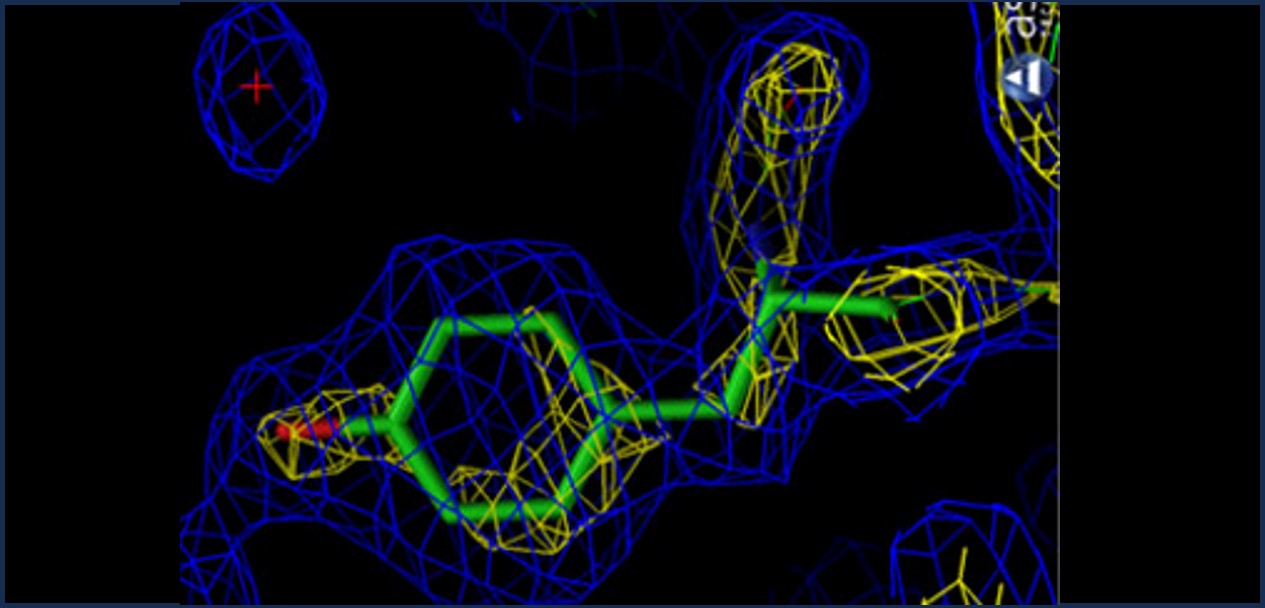
2.0 Å — Good for side chains
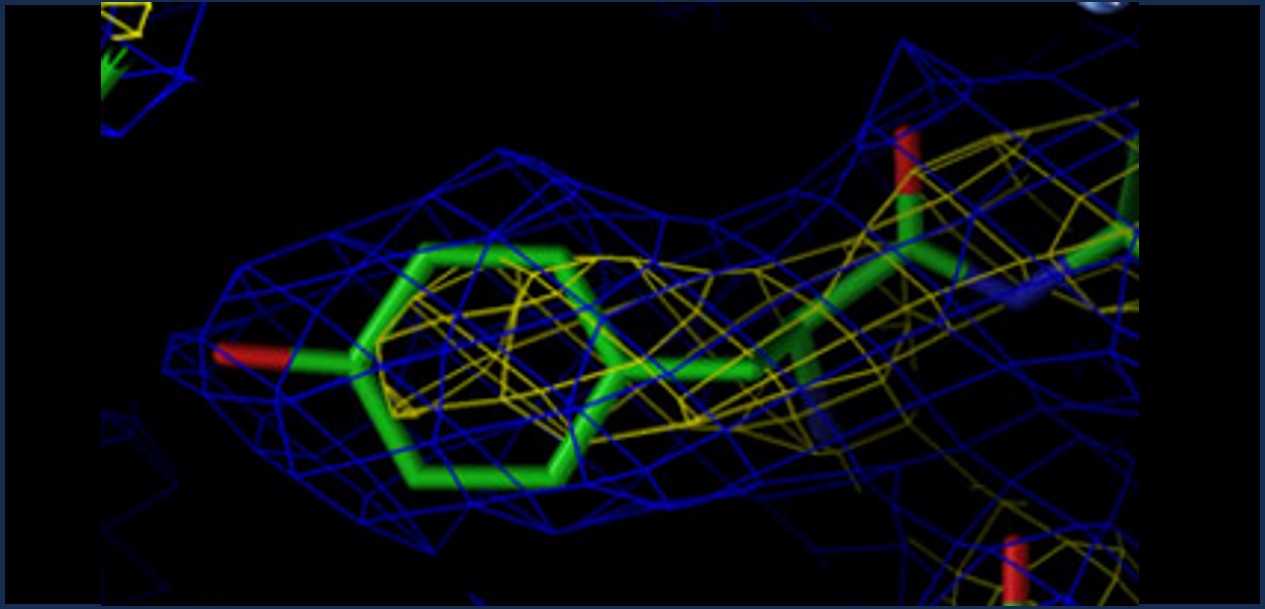
3.5 Å — Backbone visible only
| Resolution | Quality | What you can see |
|---|---|---|
| < 1.5 Å | Exceptional | Individual hydrogen atoms sometimes visible |
| 1.5–2.5 Å | Good | All heavy atoms reliable — good for drug design |
| 2.5–3.5 Å | Moderate | Backbone reliable, some side chain uncertainty |
| > 3.5 Å | Low | Use with caution — atomic positions approximate |
Ångströms: atomic distance unit
1 Ångström (Å) = 0.1 nanometres = 10-10 metres
(Flea) 1 µm
(Bacterium) 1 nm
(Protein) 1 Å
(Bond length) 0.1 Å
(Nucleus)
- A carbon–carbon bond is 1.54 Å
- A hydrogen bond is 1.8–3.5 Å
- The COX-2 binding channel is roughly 6–8 Å wide
- The ibuprofen molecule is about 8 Å across
The Drug Discovery Pipeline
Computation compresses the 15-year timeline
(2 yrs)
(weeks–months)
(1–2 yrs)
(3–4 yrs)
(6–7 yrs)
| Without computational docking | With computational docking |
|---|---|
| Synthesise drug (weeks, £10,000+) | Screen digitally (hours, nearly free) |
| Test on cells (months) | Test only the top candidates in the lab |
| >90% of candidates fail at this stage | Dramatically reduces the failure rate |
Your computational pipeline (Roadmap)
| Project | What we do | The question it answers |
|---|---|---|
| 1 | Measure Lysozyme stability; mutate | How stable is it, which amino acids matter? |
| 2 | Compare 5 crystal structures | Which structure is highest quality? |
| 3 | Dock NSAIDs into COX-2 | Which drug binds most tightly? |
| 4 | Map COX-2 binding site | Which residues touch the drug? |
| 5 | Alanine scan of binding site | Which residues are essential for binding? |
| 6 | Model resistance mutations | How do bacteria evolve to resist antibiotics? |
"By the end of Project 6, you will have run the same computational pipeline that professional drug discovery teams use."
Summary & Preparation
- A drug works by binding to a specific protein and disrupting its function.
- The protein's 3D shape determines what can bind to it.
- Good drug targets are essential, have binding pockets, and differ from healthy human proteins.
- The Protein Data Bank holds >220k real 3D structures.
- Resolution (Å) tells you how precisely a structure was determined.
- Computational docking screens thousands of drugs in hours, saving years of work.
"Think about this: if you unfold a protein — stretch it out into a straight line — does that require energy, or does energy get released? Why?"