The Binding Site and Protein-Ligand Docking
How does a drug find its target?
Lecture 3 — Shape, Chemistry and Binding Energy
The binding site is where the drug physically docks
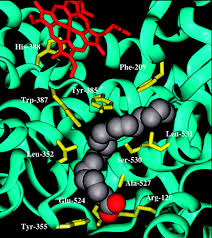
A binding site is a cavity or groove on the protein surface where a small molecule settles and interacts with surrounding amino acids.
The drug does not interact with the whole protein — only with the 10–20 amino acids that line this pocket.
Three things must match:
1. Shape — the drug must fit
2. Size — not too large, not too small
3. Chemistry — matching polarity and charge
Connect to Project 4: You will identify every amino acid within 5 Å of Ibuprofen inside COX-2 — these residues form the binding site.
Rule 1 — The drug must fit the shape of the pocket
| Situation | Result |
|---|---|
| Drug too large | Cannot enter the pocket — steric clash |
| Drug too small | Fits but makes few contacts — weak binding |
| Wrong shape, right size | Some contacts made but poor fit — moderate binding |
| Matches pocket shape precisely | Maximum contacts — strong binding ✓ |
Concrete numbers: The COX-2 binding channel is 6–8 Å wide. Ibuprofen is ~8 Å across its longest axis — a near-perfect fit. This geometric match is part of why it binds strongly.
Rule 2 — Drug VS Pocket matched chemistry
| Binding site character | Key residues | Drug should have |
|---|---|---|
| Hydrophobic pocket | Phe, Val, Ile, Leu, Met | Non-polar groups, aromatic rings |
| Polar region | Ser, Thr, Asn, Gln | H-bond donors or acceptors (-OH, -NH₂) |
| Positively charged region | Arg, Lys, His | Negatively charged group on drug |
| Negatively charged region | Asp, Glu | Positively charged group on drug |
Chemical complementarity = making sure the key is made of the right material.
A key made of butter might fit the lock perfectly in shape — but it will fail to turn it.
COX-2 — a real binding site example
COX-2 converts arachidonic acid into prostaglandins (pain and inflammation). Blocking its active site prevents this.
| Drug | Brand name | Character |
|---|---|---|
| Ibuprofen | Advil | Small, non-polar, flexible |
| Celecoxib | Celebrex | Larger, selective COX-2 inhibitor, sulfonamide group |
| Meloxicam | Mobic | Intermediate size, thiazine ring system |
Protein-ligand docking: predicting how a drug fits
What we do in Projects 3 & 4
The drug is already inside the protein — co-crystallised in the PDB structure. We are scoring a known, experimentally confirmed pose, not predicting a new one. Simpler and more reliable.
Real-world power
Screen thousands of candidates digitally in hours. Only synthesise and test the top scorers in the lab. Reduces the >90% failure rate of untargeted drug development.
Binding energy — the number that ranks drugs
| Component | Score |
|---|---|
| Complex (COX-2 + Ibuprofen) | −1,820 REU |
| Protein alone (COX-2) | −1,768 REU |
| Ibuprofen alone | −25 REU |
| Binding energy | −1,820 − (−1,768 + −25) = −27 REU |
Interpreting binding energy values
Important: Binding energy is one factor — not the only factor. Bioavailability, selectivity, and chemical stability also matter. Docking tells you about binding; the rest requires lab and clinical testing.
Structure quality affects every docking result
If the binding site geometry in your crystal structure is wrong by even 0.3 Å, your calculated binding energy will be wrong too.
| Source of error | Effect on docking |
|---|---|
| Low resolution (> 2.5 Å) | Side chain positions uncertain — binding site shape approximate |
| Missing residues | Parts of the binding site absent — contacts not calculated |
| Crystal contacts | Packing forces in the crystal may distort the binding site slightly |
The contact shell: which residues touch the drug?
| Distance | Interaction type |
|---|---|
| < 2.0 Å | Clash — atoms overlapping (problem) |
| 2.0–3.5 Å | Covalent bond or strong hydrogen bond |
| 3.5–5.0 Å | Van der Waals contact — atoms touching ✓ |
| 5.0–8.0 Å | Close neighbourhood — weak or indirect |
| > 8.0 Å | Too far to interact meaningfully |
A binding site may have 200 residues nearby, but only 12–15 are actually touching the drug. Those are the ones that matter for binding energy, drug design, and resistance mutations — and the target of Project 4.
Summary & Preparation
- A binding site is a cavity on the protein surface — shape and chemistry both determine whether binding occurs.
- Shape complementarity — the drug must physically fit the pocket geometry.
- Chemical complementarity — hydrophobic drug regions face hydrophobic residues; polar faces polar; charged faces oppositely charged.
- Binding energy = Complex score − (Protein score + Ligand score). More negative = tighter binding.
- Structure quality matters — a poor crystal structure gives unreliable docking results.
- Contact residues are within 5.0 Å of the drug — typically 12–15 residues form the functional binding site.
"In Project 5 we will mutate every binding site residue to Alanine and measure how much each mutation weakens drug binding. Think about this: if you removed the side chain of a hydrophobic residue lining the binding site, what would happen to the drug's binding energy? Why?"